20.04.2016
В 2013 г. журнал Science назвал иммунотерапию злокачественных новообразований «прорывом года». Подобное лестное определение было основано на многообещающих результатах исследования 3 фазы с ипилимумабом (Ервой, Bristol-Myers Squibb), рекомбинантным человеческим моноклональным антителом (МоА), действующим против цитотоксического антигена CTLA-4, и исследований 1 фазы с МоА к рецептору запрограммированной гибели клеток (PD-1) и его лиганду (PD-L1) [1-6]. Спустя всего лишь два года достижения в иммунотерапии опухолей не вызывали ни у кого сомнений. Прогресс был достигнут не только в лечении метастатической меланомы (мМ), где впервые были использованы анти-CTLA-4 антитела, но и в лечении более 15 видов солидных опухолей, где с успехом изучались анти-PD-1/PD-L1 антитела [7]. Более того, если по предварительным результатам по изучению анти-PD1 антител в исследованиях 1 фазы было выявлено лишь достижение значимого противоопухолевого ответа, то в исследованиях 3 фазы, проведенных у больных мМ и немелкоклеточным раком легкого (НМРЛ), было показано, что новые препараты влияют на увеличение показателя общей выживаемости [8-11]. Признанием полученных результатов является одобрение Управлением по контролю над качеством пищевых продуктов и лекарственных средств США (FDA) ниволумаба (Опдиво, Bristol-Myers Squibb) в лечении мМ, метастатического плоскоклеточного и неплоскоклеточного НМРЛ (декабрь 2014 г., март 2015 г., октябрь 2015 г. соответственно). За ниволумабом последовало одобрение пембролизумаба (Кейтруда, Merck Sharp & Dohme) в лечении мМ (сентябрь 2014 г.), а также в терапии метастатического PD-L1 позитивного НМРЛ (сентябрь 2015 г.). После того как данные МоА зарекомендовали себя в монотерапии, они стали активно изучаться в комбинациях друг с другом [12-14].
Следствием полученных положительных результатов является увеличивающееся с каждым днем число МоА, действующих на иммунные контрольные точки [15]. Как бы то ни было, опыт применения анти-CTLA-4 и анти-PD-1/PD-L1 антител внес определенные изменения в концепцию проведения клинических исследований 1 фазы с их участием. Знание подобных особенностей необходимо для успешного изучения следующего поколения подобных антител.
Появление МоА, нацеленных на контрольные молекулы, является вторым значимым событием в онкологии за последние 15 лет. Первым по праву считается появление таргетных препаратов, действующих селективно на определенные мишени молекулярных путей, вовлеченные в процессы поддержания опухолевого роста. Их изучение коренным образом изменило принципы проведения клинических исследований 1 фазы, когда исследователям пришлось уйти от существовавшего на тот момент традиционного подхода (1 препарат подходит всем пациентам с данной патологией) и начать проводить лечение с учетом молекулярных особенностей опухоли каждого отдельно взятого больного. Подобный подход позволил успешно изучить ряд новых препаратов, включая олапариб, кризотиниб и церитиниб [16-19]. Появление ингибиторов иммунных контрольных точек (ИИКТ) явилось в определенной степени вызовом для врачей, занимающихся проведением клинических исследований 1 фазы. Помимо того, что нужно было кардинальным образом изменить ставшие уже традиционными подходы к изучению лекарственных средств, их нужно было приспособить к принципиально новому классу препаратов. Смешно сказать, но появление ИИКТ заставило ряд онкологов открыть книги по иммунологии. По своей сути, новый класс препаратов представляет собой ту же самую таргетную терапию, но направленную против цитотоксических антигенов, находящихся на поверхности клеток иммунной системы или опухолевых клеток. Ответ на лечение, так же как и его продолжительность, свидетельствует о том, что подход к терапии больных данными препаратами должен сконцентрироваться на возможном развитии резистентности к ним. Одним из вариантов ее преодоления является комбинация ИИКТ с другими классами препаратов [13], которая в то же время может оказаться чрезвычайно токсичной. В связи с этим одной из причин разработки новых подходов к изучению данных препаратов является создание эффективных и малотоксичных режимов на их основе. Безусловно, подобный подход должен включать в себя рекомендации по выбору оптимальной дозы, режима и способа введения препарата, а также должен проводиться с учетом особенностей опухоли больных, иных критериев оценки эффективности проводимого лечения, так же как и выбора оптимального дизайна исследования.
Обобщая все выше изложенное, нужно сказать о том, что впечатляющие результаты, которые демонстрирует новый класс препаратов, с одной стороны, диктуют необходимость внести изменения в дизайн и концепцию проведения клинических исследований 1 фазы, а с другой, подобные «вынужденные приспособления» позволят оптимизировать проведение исследований с данными препаратами, что в конечном итоге будет способствовать скорейшему их изучению и внедрению в клиническую практику.
Особенность 1: профиль безопасности и определение максимально переносимой дозы препарата
Основной целью проведения клинических исследований 1 фазы традиционно являлось определение профиля безопасности препарата, его переносимости и рекомендуемой дозы для проведения исследования 2 фазы. Методом достижения данной цели обычно являлась эскалация дозы исследуемого препарата. При этом предполагали, что существует линейная или прямо пропорциональная зависимость между дозой препарата, его эффективностью и токсичностью. Появление ИИКТ изменило данный подход к изучению новых препаратов. В первую очередь, это связано с невозможностью определения максимально переносимой дозы (МПД) препарата, а во-вторых – с существованием большого разнообразия доз препаратов и режимов их введения, которые были ранее исследованы.
Дозолимитирующая токсичность и определение МПД
Из 13 основных клинических исследований 1 фазы по изучению анти-CTLA-4 и анти-PD-1/PD-L1 антител [3-6, 20-30] только в одном исследовании была идентифицирована дозолимитирующая токсичность (ДЛТ) препарата [24]. В большинстве исследований выбор рекомендуемой дозы препарата для проведения исследования 2 фазы исходил из максимальной вводимой дозы препарата (МВД; 10 исследований) или данных фармакокинетики (ФК; 2 исследования). В другом исследовании слишком поздно выявили ДЛТ, которую не приняли во внимание при выборе рекомендуемой дозы препарата [25]. Интересно, что для ИИКТ характерны определенные особенности развития острой и кумулятивной токсичности (за исключением анафилактических реакций, возникающих непосредственно при введении препарата). Нежелательные явления, связанные с действием препаратов, как правило, не развиваются во время первого курса терапии, а возникновение иммуно-опосредованных нежелательных явлений может произойти в достаточно отдаленные сроки. Например, независимо от степени выраженности иммуно-опосредованные нежелательные явления обычно развиваются на 8-10 неделе после начала терапии ипилимумабом [31]. В связи с тем, что ряд нежелательных явлений могут представлять угрозу для жизни пациентов, в большинстве протоколов клинических исследований оговаривается необходимость хранения препарата, на который возникла реакция, до ее разрешения до 2 степени токсичности. Несмотря на то, что формально подобные нежелательные явления не считаются ДЛТ, их расценивают как ДЛТ [34, 35]. Следовательно, традиционное определение ДЛТ, когда в расчет принимаются нежелательные явления 3-4 степени, возникшие во время первого курса терапии, неуместно в данном контексте.
Интересно, что в большинстве исследований 1 фазы с ИИКТ были использованы заранее определенные уровни эскалации дозы, включая максимально допустимую дозу. Помимо этого, в двух последних исследованиях период ДЛТ был продлен до 2 курсов [4, 30]. Несмотря на то, что для анти-CTLA-4 антител токсичность является относительно дозозависимой, величина дозы этих препаратов не коррелирует с выраженностью частоты объективного ответа (ЧОО) (таблица 1) [31].
Таблица 1. Корреляция дозы ингибиторов иммунных контрольных точек с частотой объективного ответа.
| Ингибитор иммунных контрольных точек | Доза | ЧОО (%) | Ссылка |
|---|---|---|---|
| Анти-CTLA-4 | |||
| Ипилимумаб | 0,3 мг/кг каждые 3 нед., 4 курса | 0 | Wolchok et al. [36] |
| 3 мг/кг каждые 3 нед., 4 курса | 11 | Robert et al. [10] | |
| 10 мг/кг каждые 3 нед., 4 курса; затем каждые 12 нед. в комбинации с дакарбазином |
15,2 | Robert et al. [2] | |
| Тремелимумаб | 15 мг/кг каждые 90 дней | 10,7 | Ribas et al. [37] |
| Анти-PD-1 | |||
| Пембролизумаб | 2 мг/кг каждые 3 нед. | 26 | Robert et al. [38] |
| 10 мг/кг каждые 3 нед. | 26 | ||
| 10 мг/кг каждые 2 нед. | 33,7 | Robert et al. [10] | |
| 10 мг/кг каждые 3 нед. | 32,9 | ||
| Ниволумаб | 0,1 мг/кг каждые 2 нед. | 35,3 | Topalian et al. [39] |
| 0,3 мг/кг каждые 2 нед. | 27,8 | ||
| 1 мг/кг каждые 2 нед. | 31,4 | ||
| 3 мг/кг каждые 2 нед. | 41,2 | ||
| 10 мг/кг каждые 2 нед. | 20 | ||
| 2 мг/кг каждые 3 нед. | 40 | Robert et al. [8] | |
С другой стороны, анти-PD-1/PD-L1 антитела не имеют корреляцию между дозой, эффективностью и токсичностью. Доказательством этого служит то, что ответ на терапию, так же как и степень выраженности нежелательных явлений, является одинаковым, будь то препарат, введенный в дозе 1-2 мг/кг или 20 мг/кг каждые 2 недели или каждые 3 недели [3, 4, 6, 26, 27]. Подобное полное отсутствие ДЛТ и относительно безопасный профиль токсичности ИИКТ предполагают создание альтернативных дизайнов клинических исследований [40].
Профиль безопасности ИИКТ должен кардинальным образом отличаться от действия агонистов иммунных контрольных точек. Ярким примером подобной токсичности является неудачный опыт применения агониста антитела анти-CD28 TGN1412 и развившийся при этом синдром высвобождения цитокинов [41]. Несмотря на это, по данным последних публикаций опыт безопасного применения первых агонистов моноклональных антител анти-OX4-40 и анти-CD137 в клинических исследованиях 1 фазы был успешным [42, 43].
Оптимальная доза и выбор режима введения препарата
Второй особенностью исследований 1 фазы, в которых изучаются ИИКТ, является сложность определения оптимальной дозы и выбор режима введения препарата. Из 13 известных исследований в 6 проводилось изучение препарата лишь в одной дозе [5, 20-24, 27]. В других исследованиях изучались различные режимы введения препарата, включая повторные введения его в 1, 3, 5 дни [43] или каждые 2, 3 или 4 недели вплоть до 3 месяцев лечения [33, 37]. Максимальное количество курсов при этом могло и не оговариваться.
Ярким примером может служить история изучения ипилимумаба: несмотря на то, что препарат изучался в нескольких исследованиях 1 фазы [20, 22, 23], впоследствии различные дозы препарата, так же как режимы его назначения, изучались в клинических исследованиях более поздних фаз. Например, в клиническом исследовании 2 фазы, в котором приняли участие больные мМ, проводилось сравнение между собой трех различных доз назначения ипилимумаба (0,3; 3 и 10 мг/кг [36] каждые 3 недели, 4 курса), и пациенты, заболевание которых контролировалось, могли получать поддерживающую терапию ипилимумабом каждые 12 недель. Казалось бы, что ЧОО должна возрастать с увеличением дозы препарата (0%; 4,2% и 11% для каждой группы соответственно; p=0,002), но это не было подтверждено на большей популяции больных [1, 10, 45]. Несмотря на то, что одобренной дозой ипилимумаба является 3 мг/кг каждые 3 недели [1], в одном из исследований 3 фазы проводилось изучение ипилимумаба в дозе 10 мг/кг с последующим проведением поддерживающей терапии в дозе 10 мг/кг каждые 12 недель [2]. Что касается, анти-PD-1 антител, то они неэффективны в дозе <1 мг/кг, но ЧОО при лечении ими не зависит ни от дозы (2-10 мг/кг), ни от режима введения (каждые 2 недели – каждые 3 недели) [35, 38, 46]. Комбинация ниволумаба с ипилимумабом изучалась в различных дозах (1-3 мг/кг) и режимах (последовательная или сочетанная терапия) [13, 14, 47].
Подобное огромное разнообразие режимов введения и доз ИИКТ поднимает один из наиболее сложных вопросов: должны ли они рассматриваться как «препараты таргетной терапии», которые требуют постоянного введения, или как «вакцины», для реализации эффекта которых достаточно введение 1 дозы препарата с последующим назначением ограниченного числа аналогичных доз.
Особенность 2: дизайн исследования и когорты больных
Существует большая разница между числом пациентов, включенных в первые исследования, направленные на изучение ипилимумаба (9-46 больных), и числом пациентов, включаемых в исследования сегодня, в ряде случаев превышающим тысячу больных [48]. Данные изменения являются ярким примером того, как за последние 3-5 лет изменился подход к проведению исследований 1 фазы [18, 19, 49]. Подобная стратегия позволяет сократить время от момента проведения исследований 1 фазы до регистрации бесспорно эффективных препаратов, как было, например, в случае с кризотинибом или церитинибом [18, 19]. На сегодняшний день несколько исследований 1 фазы (NCT01375842 и NCT01295827), изучающих ИИКТ, следуют по аналогичному пути.
Это несколько противоречит традиционному пути развития препарата. Основной задачей исследований 1 фазы всегда была идентификация МПД и определение дозы для проведения исследования 2 фазы. Затем в исследованиях 2 фазы проводилась оценка эффективности препарата и в случае ее выявления инициировались рандомизированные исследования 3 фазы, направленные на сравнение существующего препарата с изучаемым. История появления пембролизумаба явилась полной противоположностью, где уже в 1 фазе исследования приняли участие большие когорты больных, а целью исследования 1 фазы было выявление эффективности исследуемого препарата (рис. 1). Исследования с новыми ИИКТ следуют по аналогичному пути. Безусловно, основным преимуществом подобной тактики является сокращение времени, затрачиваемого на «становление» нового препарата. В истории с пембролизумабом FDA присвоило статус «прорывной» терапии препарату, что сократило его изучение и последующее одобрение до <4 лет [26], в то время как среднее время, затрачиваемое от начала проведения 1 фазы до одобрения, обычно составляло около 10 лет.
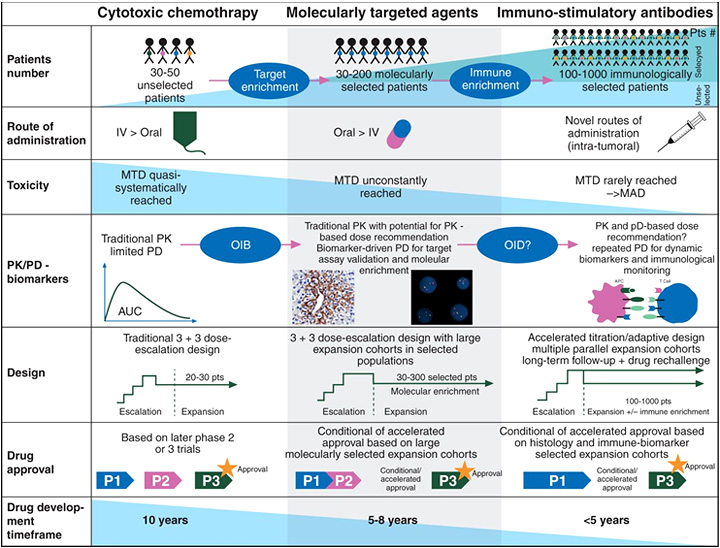
Рисунок 1. Изменение концепции проведения клинических исследований 1 фазы – от эры цитотоксических химиопрепаратов до ингибиторов ИИКТ. В исследования стали включаться большие когорты тщательно отобранных больных. Изменился способ введения и профиль безопасности препаратов – вместо максимально переносимой дозы (MTD) рассчитывают максимально вводимую дозу (MAD). Для большинства препаратов параметры фармакокинетики (PK) и фармакодинамики (PD) продолжают иметь большое значение, в частности, для определения оптимальной биологической дозы (OBD) для таргетных препаратов. По аналогии с OBD для препаратов иммунотерапии вводится понятие оптимальной иммунологической дозы (OID). Изменяется дизайн исследования: с целью раннего определения эффективности препарата ускоряется фаза эскалации дозы с последующим проведением параллельного исследования на больших когортах больных. Следствием этого является постепенная замена исследований 1 фазы на исследования 1-2 фазы и одобрение препаратов на основании результатов исследований 1 фазы. Результатом подобных изменений является сокращение времени, затрачиваемого на развитие препарата. Если раньше от момента введения исследовательского препарата человеку до регистрации препарата проходило >10 лет, то сейчас на это требуется <5 лет.
Особенность 3: отбор пациентов, биомаркеры и персонализированный подход
С идентификацией опухолеспецифических антигенов и соматических мутаций таргетный подход в терапии злокачественных новообразований вступил в эру становления персонализированной медицины и появления маркеров, предсказывающих ответ на лечение. Развитие иммуно-таргетной терапии показало, что должны быть разграничения между понятиями «стабильный» и «динамичный» биомаркер. Несмотря на опухолевую гетерогенность, такие мишени для таргетной терапии, как амплификация HER2, мутации в структуре гена EGFR, транслокация ALK, мутация BRAF, считались постоянными во времени. Экспрессия PD-L1 является более лабильной величиной, которая может быстро изменяться в течение времени в зависимости от ответа на терапию цитокинами. Несмотря на то, что позитивный статус PD-L1, как на опухолевых клетках [3, 4], так и на клетках иммунной системы [28], тесно коррелирует с ответом больных мМ и НМРЛ [9, 47], терапевтический ответ также зарегистрирован у больных, имеющих отрицательный PD-L1 статус [7, 8, 11]. Информация о таких дополнительных биомаркерах, как CTLA-4 или фракталкин, может быть весьма полезной в данной клинической ситуации [28]. Особенности изменения экспрессии PD-L1 в процессе терапии также изучались: если изменение экспрессии PD-L1 тесно коррелировало с ответом, то отсутствие подобных изменений ассоциировалось с недостаточным ответом [28]. Интересно, что корреляция между экспрессией PD-L1 и ответом на терапию анти-PD-1/PD-L1 препаратами была выявлена на образцах, ряд из которых был заготовлен за несколько лет до появления терапии ИИКТ [4]. Иммунологический мониторинг таких биомаркеров, как ИЛ-18, индуцированный интерфероном Т-клеточный альфа хемоаттрактант, ИНФ-γ и активированный Т-клеточный CD8+, не выявил подобной корреляции.
Особенность 4: критерии участия в исследовании
Учитывая специфический профиль безопасности ИИКТ и наметившуюся стратегию развития данных препаратов, возникает вопрос, насколько строго мы должны придерживаться существующих критериев для участия пациентов в клинических исследованиях 1 фазы, направленных на их изучение. Безусловно, данные препараты являются менее токсичными, чем цитотоксические химиопрепараты (рис. 1). В традиционных дизайнах клинических исследований 1 фазы пациенты, имеющие статус по шкале ECOG ≥2, высокий уровень ЛДГ, низкий уровень альбумина, большое количество очагов метастатического поражения или метастазы в головном мозге, не могли быть включены в исследование [51-53]. Напротив, у больных с метастатическим поражением головного мозга терапия ИИКТ позволяла получить объективный ответ и улучшение клинических симптомов заболевания без нарастания токсичности [54, 55]. Аналогичным образом, не было отмечено дополнительной токсичности у пациентов с высоким уровнем ЛДГ, получающих тремелимумаб [25], участников исследования со статусом по шкале ECOG 2, находящихся на терапии BMS-936559 [4], или у больных уротелиальным раком мочевого пузыря с наличием неблагоприятных факторов прогноза и получающих атезолизумаб [29]. В связи с этим было бы логичным расширить критерии включения для участия пациентов в клинических исследованиях 1 фазы, направленных на изучение ИИКТ. Следует оговориться, что разумнее это было бы делать после четкого определения профиля безопасности исследуемого препарата после прохождения его через фазу эскалации дозы и одной большой когорты больных. Подобный подход позволит не только идентифицировать дозу препарата, но и ускорит его развитие.
Особенность 5: фармакокинетика и фармакодинамика
Фармакокинетика
По своей сути, клинически активные ИИКТ являются иммуноглобулинами (Ig или антителами). Они состоят из двух высокоспецифичных антиген-связывающих фрагментов (Fab – fragment of antigen binding) и одного фрагмента, способного к кристаллизации (Fc – fragment crystallizable). В зависимости от строения, аминокислотного состава тяжелых цепей и выполняемых ими эффекторных функций выделяют пять классов Ig (IgА, IgD, IgE, IgG, IgM). Основными Ig сыворотки крови человека являются IgG, поэтому все одобренные антитела, так же как ИИКТ, относятся к данному классу. Единое происхождение объясняет схожий ФК профиль между отдельными ИИКТ, в том числе дозозависимую максимальную концентрацию препарата в плазме крови (Cmax) и площадь под кривой «концентрация-время» (AUC), а также медиану периода полураспада 16 дней (9-21 дней). Тем не менее, существуют и различия: если большинство ИИКТ являются IgG1 (ипилимумаб, атезолизумаб или дувалумаб), то некоторые из них относятся к IgG2 или IgG4 (тремелимумаб и ниволумаб соответственно). Подобные различия определяют биоактивность препаратов: IgG1 и IgG3 оказывают антителозависимую клеточно-опосредованную цитотоксичность, тогда как IgG4 более задействован в альтернативных путях активации комплемента.
Помимо этого, ФК профиль антител является более сложным, чем у малых молекул, и характеризуется определенными особенностями процессов абсорбции, распределения и элиминации [56]. Например, ограничение растворимости препаратов ведет к ряду технических вопросов, касающихся введения их только внутривенно, внутримышечно, подкожно или непосредственно в опухолевый очаг и невозможности их перорального назначения. Помимо этого, такие особенности, как «селективная элиминация», могут привести к неправильной оценке распределения препарата в стабильном состоянии [57]. Более того, такие особенности, как рецепторно-опосредованный эндоцитоз, дозозависимый период полураспада, рециркуляция рецепторов и влияние циркулирующих растворимых форм таргетных молекул (растворимый PD-L1), определяют сложность ФК IgG [56, 58, 59]. Все вышеперечисленные параметры приводят к различию между ФК профилем отдельных ИИКТ, что отражается на активности препаратов и, в конечном счете, выживаемости пациентов [60, 61]. И, наконец, несмотря на то, что большинство ИИКТ представляют собой полностью человеческие антитела, описано несколько случаев существования нечеловеческих МоА [30]. Последнее необходимо принимать во внимание не только при тщательном мониторировании участников исследования 1 фазы, но и при выборе рекомендуемой дозы препарата, режима и способа его введения.
Фармакодинамика
Данные по фармакодинамике (ФД) представлены лишь в ограниченном количестве исследований. В исследованиях, направленных на изучение ниволумаба [3] и анти-PD-L1 BMS-936559 [4], медиана связывания PD-1 и PD-L1 рецептора составила 64-70%. Интересно то, что она была идентичной как для анти-PD1/PD-L1 молекул, так и для доз в диапазоне от 0,1 до 10 мг/кг. Иммунологический мониторинг, включая CD4+ и CD8+ T-клеточный мониторинг, был также выполнен лишь в ряде исследований, так же как определение повторных доз цитокинов врожденного или приобретенного иммунного ответа [29]. Подобные оценки иммунологических показателей в процессе лечения следует более широко использовать в связи с тем, что они не только могут являться потенциальными биомаркерами иммунного ответа, но и являться критериями лекарственного ответа клеток иммунной системы. Способны ли другие показатели, действующие на ФД таких зарегистрированных антител, как, например, ритуксимаб [62], аналогичным образом влиять на ФД ИИКТ, пока остается предметом обсуждения. В ряде недавно опубликованных работ было показано, что некоторые фракции FcγR являются критичными для функционирования нескольких изотипов ИИКТ [63-65]. В связи с этим те знания и опыт, которые были приобретены при изучении особенностей показателей ФД зарегистрированных антител, должны быть использованы для изучения ИИКТ.
Обобщая вышесказанное, необходимо отметить, что существует разительный контраст между тем незначительным объемом знаний о ФК и ФД ИИКТ, который представлен в результатах исследований 1 фазы, и тем объемом информации, который только предстоит изучить. Это касается механизмов антитело-зависимой клеточной токсичности, ее динамики в зависимости от изотипа антитела, наличие комплемента и экспрессии мишени, взаимодействие между иммунными эффекторными клетками. Более глубокое понимание данной информации представляет большой интерес.
Особенность 6: оценка эффективности проводимого лечения
На сегодняшний день критерии оценки ответа на терапию ИИКТ остаются предметом оживленных дискуссий. В ранних исследованиях 1 фазы по изучению ИИКТ традиционно использовали модифицированные критерии, разработанные Всемирной Организацией Здравоохранения (мВОЗ) или критерии RECIST 1.0/1.1 (при изучении ипилимумаба и анти-PD1/PD-L1 антител соответственно). С целью оценки того разнообразия ответов, которое может быть связано с действием ИИКТ, совсем недавно был введен критерий оценки иммунного ответа [66]. Интересные нововведения, касающиеся особенности оценки иммунного ответа, были предложены для больных мМ, получающих ипилимумаб. Подобные изменения включают в себя редукцию числа таргетных очагов и одномерных измерений [67, 68]. Должны ли подобные особенности учитываться при оценке ответа на терапию другими ИИКТ или при других видах злокачественных новообразований?! Более того, интересно, что при оценке ответа на терапию ИИКТ был описан ряд необычных типов ответа, таких как диссоциированный ответ, отсроченный ответ и псевдопрогрессия, механизмы возникновения которых остаются неясными. Несомненно, при терапии данными препаратами может возникнуть псевдопрогрессия заболевания, могут появиться новые нетаргетные очаги, но до сих пор остается невозможным охватить весь спектр вариантов ответа, которые бывают при лечении ими. Необходимо внести ряд изменений, в том числе касающихся особенности статистического анализа, оценки КТ-снимков, метаболической визуализации и маркеров иммунного ответа. В связи с тем, что время развития ответа широко варьирует, начиная от 6 недель до нескольких месяцев после начала лечения, или ответ развивается после прекращения лечения, должны быть введены такие понятия, как скорость контроля заболевания и скорость роста опухоли [69, 70]. Более того, такие альтернативные подходы, как метаболическая визуализация или иммунологический мониторинг, могут оказаться весьма ценными, особенно в самом начале изучения препарата – при проведении исследований 1 фазы. В конце концов, традиционные подходы терапии, проводимые при прогрессировании заболевания после терапии ИИКТ, могут вызвать совершенно неожиданные ответы со стороны опухоли [71, 72]. Подобные отсроченные ответы приводят к тому, что появляется расхождение в понятиях выживаемость без прогрессирования и общая выживаемость у пациентов, получающих терапию ИИКТ. Сегодня это не всегда учитывается при оценке эффекта во время проведения исследований 1 фазы.
Возможные будущие особенности
На сегодняшний день преодоление первичной и вторичной резистентности к терапии ИИКТ является первостепеннейшей задачей современной иммуноонкологии. Комбинация существующих препаратов, так же как модернизация способов их введения, является одним из вариантов решения данной проблемы. Подобные комбинации могут потребовать изменения доз препаратов или режима их введения. Следует предвидеть, что в большинстве случаев при применении комбинации двух ИИКТ могут возникнуть различного вида серьезные нежелательные явления, как, например, было в случае комбинации ипилимумаба с ниволумабом [13, 14, 47]. Подобным же образом комбинация одного ИИКТ с любым другим противоопухолевым препаратом ставит вопрос оптимальной последовательности назначения, доз и режима введения препаратов. Например, исследование 1 фазы по оценке эффективности комбинации ипилимумаба с вемурафенибом [73] было досрочно прекращено в связи с высокой (III-IV ст.) частотой печеночной токсичности (у 4 и 3 из 6 пациентов соответственно), несмотря на то, что она была бессимптомна и обратима. В исследовании 1 фазы, направленном на изучение комбинации ипилимумаба с ниволумабом [14], также была выявлена высокая частота нежелательных явлений (III-IV ст.) и ДЛТ (53% и 21% соответственно), но данные нежелательные явления были контролируемыми и их спектр соответствовал аналогичному, наблюдаемому при применении препаратов в монорежиме. Комбинации с неиммунологическими противоопухолевыми препаратами также должны быть тщательно продуманы. В подобных исследованиях наиболее активный препарат должен назначаться в максимально эффективной дозировке, тогда как доза экспериментального препарата должна исходить из его ФК, ФД особенностей (введение с интервалами максимальных доз препаратов, чья активность зависит от достижения максимальной концентрации, и непрерывное введение в низких дозах препаратов, действие которых достигается с течением времени). При этом во всех протоколах исследования, направленных на изучение комбинаций этих препаратов, должна содержаться информация об особенностях снижения дозы или длительных перерывах в лечении.
Интересно, что при терапии ИИКТ высокую эффективность при минимальной токсичности можно достигнуть за счет способов введения препарата, например, вводя препарат локально инъекционно. Этот подход активно изучается в исследованиях 1 фазы [74]. Как бы то ни было, данная область еще требует дальнейшего изучения и развития.
Таким образом, современная терапия злокачественных новообразований вступила в новую эру – эру иммуноонкологии, с целью более успешного развития которой необходимо изменить существующие или создать новые принципы проведения клинических исследований.
Литература: